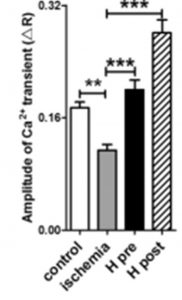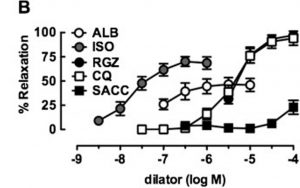
Albuterol (ALB) in comparison with a novel bronchodilator (RGZ) in the relaxation of isolated mouse airway tissue. From: Donovan C, Simoons M, Esposito J, Ni Cheong J, Fitzpatrick M, Bourke JE. Rosiglitazone is a superior bronchodilator compared to chloroquine and beta-adrenoceptor agonists in mouse lung slices. Respiratory research. 2014;15:29, reproduced under the terms of a Creative Commons Attribution 4.0 International License
Albuterol is historically linked to bronchodilation, as it activates the beta-2 subtype of the adrenergic receptor (β2-AR). It is available as two discrete isomers; S-albuterol and R-albuterol (also known as albuterol). A treatment consisting of a mixture of these isomers may have a complementary effect on therapeutic corticosteroids and a beneficial effect on inflammation, but purified S-albuterol lacks this property
1. Albuterol elicits generally beneficial effects in lung tissue through the modulation of different ions, occurring as a result of β2-AR activation. However, these same effects may precipitate into adverse events when albuterol is introduced into different tissues that also possess adrenergic receptors.
Albuterol and Cardiovascular Effects
High-dose albuterol is associated with increased risks of tachycardia and tachypnea (accelerated heart rate and breathing) and with ventricular arrythmia. This may be associated with sharp decreases in cellular potassium as a result of β2-AR activation, which may lead to increased weakness in various muscle tissue types
2. These effects may result in acute myocardial injury in severe cases
2. On the other hand, albuterol may also increase intracellular calcium in these cells, which may be associated with increased contractility in cardiac muscle
3.
Albuterol and Respiratory Conditions
Treatment with albuterol is a well-established method in the alleviation of conditions such as asthma. Albuterol at concentrations of 10
7M to 10
6M significantly reduced the contraction in isolated guinea pig trachea mediated by 10
7M to 10
3M insulin
4. The administration of this compound may also address conditions such as acute respiratory distress syndrome (ARDS) and other acute lung conditions through the regulation of sodium/potassium-ATPase (Na/K-ATPase)
5. This enzyme is involved in the reduction of Na
+ concentrations in alveolar spaces, thus contributing to the control of fluid accumulation and swelling in these areas of lung tissue
5. Recent research has improved the understanding of how albuterol regulates this ATPase. A study using rat alveolar cells demonstrated the ability of the molecule to induce the influx of intracellular calcium through calcium release-activated calcium (CRAC) channels. This in turn enhances the aggregation of Na/K-ATPase at the plasma membrane of alveolar cells, which is also mediated by β2-AR activation
5.
Albuterol may also be useful in trials that determine the genes that may be involved in respiratory function and health (e.g. airway responsiveness or the regulation of inflammation). The administration of albuterol failed to change lung resistance in mice with an extra copy of the
Plp gene, although it had a negative effect on this measure in normal and carrier mice
6. This indicates a role for the gene in responsiveness. Similarly, albuterol may also be used in the study of conditions that may be associated with increased risks of respiratory dysfunction. For example, congenital cryptorchidism (or retention of the testicles in the abdominal cavity) may be comorbid with asthma symptoms in some species
7. Treatment with albuterol significantly reduced methacholine-resistance in both rats with this condition and corresponding control animals
7. This treatment also resulted in the increased down-regulation of interleukin-4 and -6 in the lungs of rats affected by cryptorchidism
7. This indicates a role for albuterol in the control of respiratory inflammation for animals with co-morbid conditions. Albuterol may also be used in trials assessing novel treatments for airway constriction and other respiratory symptoms
8.
References:
1. Ameredes BT, Calhoun WJ. Levalbuterol versus albuterol.
Current allergy and asthma reports. 2009;9(5):401-409.
2. Matos J, Jenni S, Fischer N, Bienz H, Glaus T. [Myocardial damage and paroxysmal ventricular tachycardia in a dog after Albuterol intoxication].
Schweizer Archiv fur Tierheilkunde. 2012;154(7):302-305.
3. Ogrodnik J, Niggli E. Increased Ca(2+) leak and spatiotemporal coherence of Ca(2+) release in cardiomyocytes during beta-adrenergic stimulation.
The Journal of physiology. 2010;588(Pt 1):225-242.
4. Sharif M, Khan BT, Ajmal K, Anwar MA. Acute effect of insulin on guinea pig airways and its amelioration by pre-treatment with salbutamol.
JPMA. The Journal of the Pakistan Medical Association. 2014;64(8):932-935.
5. Keller MJ, Lecuona E, Prakriya M, et al. Calcium release-activated calcium (CRAC) channels mediate the beta(2)-adrenergic regulation of Na,K-ATPase.
FEBS letters. 2014;588(24):4686-4693.
6. Rodriguez E, Sakowski L, Hobson GM, et al. Plp1 gene duplication inhibits airway responsiveness and induces lung inflammation.
Pulmonary pharmacology & therapeutics. 2015;30:22-31.
7. Rodriguez E, Barthold JS, Kreiger PA, et al. The orl rat is more responsive to methacholine challenge than wild type.
Pulmonary pharmacology & therapeutics. 2014;29(2):199-208.
8. Donovan C, Simoons M, Esposito J, Ni Cheong J, Fitzpatrick M, Bourke JE. Rosiglitazone is a superior bronchodilator compared to chloroquine and beta-adrenoceptor agonists in mouse lung slices.
Respiratory research. 2014;15:29.